

Stwardnienie zanikowe boczne (ALS, choroba Charcota, choroba motorycznego neuronu ruchowego, choroba Lou Geringa, postępująca atrofia mięśniowa) jest śmiertelną chorobą ośrodkowego układu nerwowego charakteryzującą się selektywną defekacją motoryczną neurony. Klinicznie objawia się on narastającym osłabieniem mięśni i wyszczuplaniem mięśni, który pochłania coraz więcej macierzy mięśniowych. W końcu prowadzi to do całkowitego unieruchomienia pacjenta i naruszenia oddychania. Choroba kończy się w śmiertelnym przypadku średnio przez 5 lat od rozpoznania. Tylko 7% pacjentów żyje dłużej niż 60 miesięcy.
Czołowi naukowcy zajmujący się medycyną nie do końca zrozumieli przyczyny tej choroby, a mechanizm rozwoju nie został dokładnie zbadany. Opowiedz o tym, co jest znane nauce.
Spis treści
- 1Informacje ogólne
- 2Przyczyny
- 3Mechanizm rozwoju
Informacje ogólne
Choroba jest znana nie tak dawno temu. Został po raz pierwszy opisany przez Jean-Martina Charcota w 1869 roku. Według statystyk wykryto 2-5 osób na 100 000 populacji rocznie, co wskazuje, że ta patologia jest stosunkowo rzadka. W ostatnich latach obserwuje się tendencję do wzrostu chorobowości i "odmłodzenia" ALS. W sumie jest około 70 tysięcy pacjentów ze stwardnieniem zanikowym bocznym. Mężczyźni cierpią nieco częściej niż kobiety, stosunek ten wynosi: 1, ale po 65 latach stosunek ten jest wyrównany. Zazwyczaj choroba objawia się u osób w wieku powyżej 50 lat. Niedawno wyrażono opinię, że przypadki stwardnienia zanikowego bocznego są częściej rejestrowane w sposób wysoce inteligentny ludzie, profesjonaliści w swojej branży, a także sportowcy, sportowcy, którzy przez całe życie wyróżniali się silnym zdrowiem.
Przyczyny
 Rozwój choroby wiąże się z defektem enzymu, który chroni komórki przed niszczącym działaniem wolnych rodników.
Rozwój choroby wiąże się z defektem enzymu, który chroni komórki przed niszczącym działaniem wolnych rodników.Pomimo aktywnych badań w tym kierunku, przyczyna rozwoju ALS wciąż nie jest wymieniona. Większość naukowców ma tendencję do myślenia o genetycznej chorobie, tj. Oczekuje się kilku efektów.
Zakłada się, że główna rola należy do zmiany właściwości dysmutazy ponadtlenkowej-1. Jest to enzym, który chroni komórki organizmu przed zniszczeniem pod wpływem rodników tlenowych, tj. ma funkcję antyoksydacyjną. U 30% pacjentów ze stwardnieniem zanikowym bocznym wykrywa się mutacje w 21 genach chromosomu kodujących dysmutazę ponadtlenkową-1. W 25% przypadków mutacja ta jest przekazywana przez dziedziczenie kolejnym pokoleniom, u pozostałych pacjentów rozwija się po raz pierwszy w rodzaju pod wpływem niekorzystnych czynników. Być może istotną rolę odgrywają także mutacje innych struktur: neurofile (formacje tworzące szkielet komórki nerwowej, nadające mu formę), białko związane z pęcherzykami.
Wśród wielu czynników, które mogą powodować rozwój ALS, szczególną uwagę zwraca się na:
- exytotoksyczność glutaminianu - uruchomienie uszkodzenia neuronów pod wpływem glutaminianu (substancji przenoszącej "informację" w układzie nerwowym);
- nadmierne spożycie jonów wapnia w komórce, naruszenie związku pomiędzy wapniem wewnątrzkomórkowym i pozakomórkowym;
- brak czynników neurotroficznych;
- procesy autoimmunologiczne;
- egzotoksyny (patologiczny wpływ pestycydów rolniczych, ołowiu);
- palenie.
Przekonujące dane na temat konkretnych skutków któregokolwiek z powyższych czynników nie są jeszcze dostępne, zakłada się ich wspólną rolę w rozwoju choroby.
Dlaczego stwardnienie zanikowe boczne wpływa tylko na neurony ruchowe, nadal pozostaje zagadką.
Mechanizm rozwoju
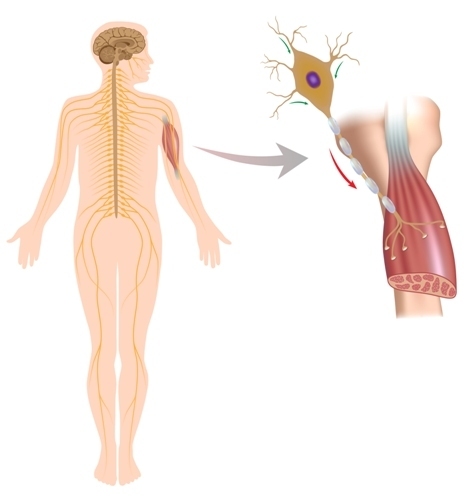
Istota choroby polega na degeneracji neuronów ruchowych, tj. pod wpływem wielu przyczyn wyzwalany jest proces niszczenia komórek nerwowych odpowiedzialnych za skurcze mięśni. Proces ten wpływa na neurony kory mózgowej, jądra mózgu i neurony przednich rogów rdzenia kręgowego. Neurony motoryczne umierają, a nikt inny nie spełnia ich funkcji. Impulsy nerwowe do komórek mięśniowych już nie nadchodzą. W wyniku osłabienia mięśni, niedowładu i porażenia dochodzi do atrofii tkanki mięśniowej. Jak to się dzieje?
Jeśli podstawą stwardnienia zanikowego bocznego jest mutacja w genie dysmutazy-superutlenku-1, wówczas proces wygląda tak. Zmutowana dysmutaza ponadtlenkowa-1 akumuluje się w mitochondriach neuronów motorycznych (w elektrolizerach komórki). To "zakłóca" normalny transport wewnątrzkomórkowy, w tym ruch formacji białkowych. Białka łączą się ze sobą, ponieważ zostały sklejone, co powoduje proces degeneracji komórek.
Jeśli przyczyną jest nadmiar glutaminianu, wówczas mechanizm wyzwalający destrukcję neuronów ruchowych wygląda następująco: glutaminian otwiera kanały w błonie neuronów dla wapnia. Wapń wpada do komórek. Nadmiar wapnia z kolei aktywuje enzymy wewnątrzkomórkowe. Enzymy wydają się "trawić" struktury komórek nerwowych, podczas gdy powstaje duża liczba wolnych rodników. A te wolne rodniki uszkadzają neurony, stopniowo prowadząc do ich całkowitego zniszczenia.
Przyjmuje się, że rola innych czynników w rozwoju ALS polega również na inicjacji utleniania wolnych rodników.
Jeśli podsumujemy wszystkie powyższe, wówczas staje się jasne, że w badaniu przyczyn i mechanizmu rozwoju stwardnienia zanikowego bocznego bardziej nieznane niż badane. Dwuznaczność tych pytań znajduje odzwierciedlenie w sposobach leczenia tej choroby. Leczenie, objawy i rozpoznanie tej choroby można znaleźć w poniższym artykule.
Program edukacyjny dotyczący neurologii, prezentacja wideo na temat "Stwardnienie zanikowe boczne

Obejrzyj ten film na YouTube


