

Amyotrofisk lateralsklerose (ALS, Charcot's sykdom, motor motoneuronsykdom, Lou Gering's sykdom, progressiv muskelatrofi) er en dødelig sykdom i sentralnervesystemet kjennetegnet ved selektivt nederlag av motor nevroner. Klinisk manifesterer seg seg som en økende muskel svakhet og muskelslanking, noe som sprenker flere og flere muskelarrayer. Til slutt fører dette til en fullstendig immobilitet av pasienten og et brudd på pusten. Sykdommen avsluttes i dødelig tilfelle i gjennomsnitt i 5 år etter diagnosen. Bare 7% av pasientene lever mer enn 60 måneder.
De ledende medisinske forskerne har ennå ikke fullt ut forstått årsakene til denne sykdommen, og utviklingsmekanismen er ikke grundig studert. Fortell deg hva som er kjent for vitenskapen.
innhold
- 1Generell informasjon
- 2årsaker til
- 3Mekanismen for utvikling
Generell informasjon
Sykdommen er kjent ikke så lenge siden. Det ble først beskrevet av Jean-Martin Charcot i 1869. Ifølge statistikken oppdages 2-5 personer per 100 000 individer i året, noe som indikerer at denne patologien er relativt sjelden. I de senere år har det vært en trend mot en økning i sykelighet og "foryngelse" av ALS. Totalt er det ca 70 000 pasienter med amyotrofisk lateral sklerose. Menn lider noe oftere enn kvinner, forholdet er: 1, men etter 65 år er dette forholdet utlignet. Vanligvis manifesterer sykdommen seg hos mennesker eldre enn 50 år. Mer nylig ble uttalelsen uttrykt at tilfeller av amyotrofisk lateralsklerose blir registrert hyppigere folk, fagfolk i deres virksomhet, samt idrettsutøvere, idrettsutøvere som gjennom hele livet var preget av sterk helse.
årsaker til
 Utviklingen av sykdommen er forbundet med en defekt i enzymet som beskytter celler mot skadelige effekter av frie radikaler.
Utviklingen av sykdommen er forbundet med en defekt i enzymet som beskytter celler mot skadelige effekter av frie radikaler.Til tross for aktiv forskning i denne retningen, er årsaken til utviklingen av ALS fortsatt ikke navngitt. Hovedparten av forskerne har en tendens til å tenke på den polyfaktoriale genetese av sykdommen, dvs. Flere effekter er forventet.
Det antas at hovedrolle tilhører forandringen i egenskapene av superoksiddismutase-1. Det er et enzym som beskytter kroppens celler mot ødeleggelse under påvirkning av oksygenradikaler, dvs. har en antioksidant funksjon. Hos 30% av pasientene med amyotrofisk lateralsklerose detekteres mutasjoner i 21 kromosomgener som koder for superoksiddismutase-1. Og i 25% av tilfellene overføres denne mutasjonen ved arv til de neste generasjonene, i de gjenværende pasientene utvikler den for første gang i slekten under påvirkning av ugunstige faktorer. Kanskje, mutasjoner av andre strukturer spiller også en rolle: neurofilamenter (formasjonene som gir rammen av nervecellen, gir den en form), det vesikkelassosierte proteinet.
Blant de mange faktorene som kan forårsake utviklingen av ALS, er det spesielt oppmerksom på:
- glutamateksytotoksisitet - lansering av skade på nevroner under påvirkning av glutamat (et stoff som overfører "informasjon" i nervesystemet);
- overdreven inntak av kalsiumioner inne i cellen, et brudd på forholdet mellom intracellulært og ekstracellulært kalsium;
- mangel på nevrotrofiske faktorer;
- autoimmune prosesser;
- eksotoksiner (patologisk effekt av landbruksmidler, bly);
- røyke.
Overbevisende data om de spesifikke effektene av noen av de ovennevnte faktorene er ikke tilgjengelig ennå, deres felles rolle i utviklingen av sykdommen antas.
Hvorfor amyotrofisk lateral sklerose bare påvirker motorneuroner, er fortsatt et mysterium.
Mekanismen for utvikling
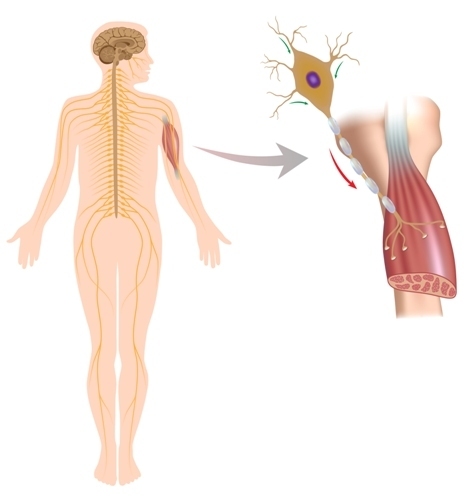
Essensen av sykdommen ligger i degenerasjonen av motorneuroner, dvs. Under påvirkning av en rekke årsaker utløses prosessen med ødeleggelse av nerveceller som er ansvarlig for muskelkontraksjoner. Denne prosessen påvirker nevronene i hjernebarken, kjernen i hjernen og nevronene i de fremre hornene i ryggmargen. Motorneuroner er døende, og ingen andre utfører sine funksjoner. Nerveimpulser til muskelceller kommer ikke lenger. Og muskler svekkes, parese og lammelse utvikles, utvikler muskelvevatrofi. Hvordan skjer dette?
Hvis grunnlaget for amyotrofisk lateralsklerose er en mutasjon i gen-superoksyd dismutasen-1, ser prosessen slik ut. Mutant superoksyd dismutase-1 akkumuleres i mitokondrier av motorneuroner (i kraftverkene i cellen). Dette "interfererer" med normal intracellulær transport, inkludert bevegelsen av proteinformasjoner. Proteiner kombinerer med hverandre, som det var sammen, og dette utløser prosessen med celledegenerering.
Hvis årsaken er et overskudd av glutamat, ser mekanismen for å utløse ødeleggelsen av motorneuroner slik ut: glutamat åpner kanaler i membranen til nevroner for kalsium. Kalsium rushes inn i cellene. For mye kalsium aktiverer i sin tur intracellulære enzymer. Enzymer synes å "fordøye" strukturer av nerveceller, mens et stort antall frie radikaler dannes. Og disse frie radikaler ødelegger nevroner, som gradvis fører til fullstendig ødeleggelse.
Det antas at rollen til andre faktorer i utviklingen av ALS også ligger i initiering av friradikaloksydasjon.
Hvis vi oppsummerer alt ovenfor, blir det klart at i studien av årsakene og mekanismen for utvikling av amyotrofisk lateralsklerose mer ukjent enn den studerte. Tvetydigheten i disse spørsmålene reflekteres i måtene å behandle denne sykdommen. Behandling, symptomer og diagnose av denne sykdommen finner du i følgende artikkel.
Utdannelsesprogram for nevrologi, video presentasjon på temaet "Amyotrofisk lateral sklerose

Se denne videoen på YouTube

