

Amyotrofische laterale sclerose (ALS, de ziekte van Charcot, motoneuronziekte, ziekte van Lou Gering, progressief spieratrofie) is een dodelijke ziekte van het centrale zenuwstelsel die wordt gekenmerkt door het selectief verslaan van de motor neuronen. Klinisch gezien manifesteert het zich als een toenemende spierzwakte en spiervermindering, die steeds meer spierarrays omvat. Uiteindelijk leidt dit tot een volledige onbeweeglijkheid van de patiënt en een schending van de ademhaling. De ziekte eindigt in het dodelijke geval gemiddeld 5 jaar na de diagnose. Slechts 7% van de patiënten leeft meer dan 60 maanden.
De leidende medische wetenschappers hebben de oorzaken van deze ziekte nog niet volledig begrepen en het ontwikkelingsmechanisme is nog niet grondig bestudeerd. Vertel je wat de wetenschap bekend is.
inhoud
- 1Algemene informatie
- 2oorzaken van
- 3Het mechanisme van ontwikkeling
Algemene informatie
De ziekte is nog niet zo lang geleden bekend. Het werd voor het eerst beschreven door Jean-Martin Charcot in 1869. Volgens de statistieken worden 2-5 personen per 100 000 inwoners per jaar gedetecteerd, wat erop wijst dat deze pathologie relatief zeldzaam is. In de afgelopen jaren is er een trend geweest naar een toename van morbiditeit en "verjonging" van ALS. In totaal zijn er ongeveer 70 duizend patiënten met amyotrofische laterale sclerose. Mannen lijden iets vaker dan vrouwen, de ratio is 1, maar na 65 jaar is deze verhouding geëgaliseerd. Meestal manifesteert de ziekte zich bij mensen ouder dan 50 jaar. Meer recent werd de mening uitgesproken dat gevallen van amyotrofische laterale sclerose vaker worden geregistreerd in zeer intelligent mensen, professionals in hun bedrijf, maar ook atleten, atleten die zich gedurende hun hele leven onderscheiden door een sterke gezondheid.
oorzaken van
 De ontwikkeling van de ziekte is geassocieerd met een defect in het enzym dat cellen beschermt tegen de schadelijke effecten van vrije radicalen.
De ontwikkeling van de ziekte is geassocieerd met een defect in het enzym dat cellen beschermt tegen de schadelijke effecten van vrije radicalen.Ondanks actief onderzoek in deze richting, wordt de oorzaak van de ontwikkeling van ALS nog steeds niet genoemd. Het grootste deel van de wetenschappers denkt vaak na over de polyfactoriële genese van de ziekte, d.w.z. Er worden verschillende effecten verwacht.
Er wordt verondersteld dat de belangrijkste rol tot de verandering in de eigenschappen van superoxide dismutase-1 behoort. Het is een enzym dat lichaamscellen beschermt tegen vernietiging onder invloed van zuurstofradicalen, d.w.z. heeft een antioxidantfunctie. Bij 30% van de patiënten met amyotrofische laterale sclerose worden mutaties in 21 chromosoomgenen die coderen voor superoxide-dismutase-1 gedetecteerd. En in 25% van de gevallen wordt deze mutatie overgedragen door overerving aan de volgende generaties, in de resterende patiënten ontwikkelt het zich voor de eerste keer in het geslacht onder invloed van ongunstige factoren. Misschien spelen mutaties van andere structuren ook een rol: neurofilamenten (de formaties die het raamwerk van de zenuwcel vormen, waardoor het een vorm krijgt), het vesikel-geassocieerde eiwit.
Onder de vele factoren die de ontwikkeling van ALS kunnen veroorzaken, wordt speciale aandacht besteed aan:
- glutamaat-exytotoxiciteit - de lancering van schade aan neuronen onder invloed van glutamaat (een stof die "informatie" in het zenuwstelsel overdraagt);
- overmatige inname van calciumionen in de cel, een schending van de relatie tussen intracellulair en extracellulair calcium;
- een gebrek aan neurotrofische factoren;
- auto-immuunprocessen;
- exotoxinen (pathologisch effect van landbouwpesticiden, lood);
- roken.
Overtuigende gegevens over de specifieke effecten van een van de bovengenoemde factoren zijn nog niet beschikbaar, hun gezamenlijke rol in de ontwikkeling van de ziekte wordt verondersteld.
Waarom amyotrofische laterale sclerose alleen motorische neuronen beïnvloedt, is nog steeds een mysterie.
Het mechanisme van ontwikkeling
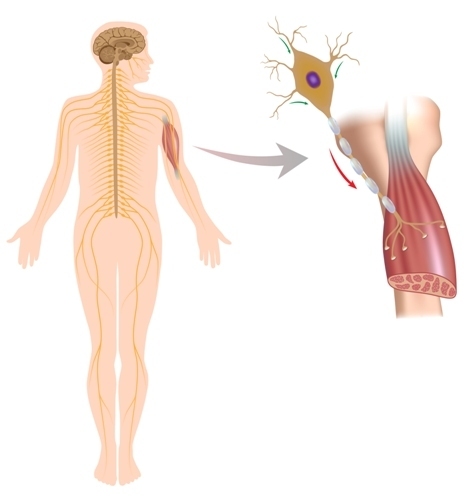
De essentie van de ziekte ligt in de degeneratie van motorneuronen, d.w.z. onder invloed van een aantal redenen wordt het proces van vernietiging van zenuwcellen die verantwoordelijk zijn voor spiersamentrekkingen geactiveerd. Dit proces beïnvloedt de neuronen van de hersenschors, de kernen van de hersenen en de neuronen van de voorhoorns van het ruggenmerg. Motorneuronen sterven en niemand anders voert zijn functies uit. Zenuwimpulsen naar spiercellen komen niet meer. En spieren verzwakken, parese en verlamming ontwikkelen zich, spierweefselatrofie ontwikkelt zich. Hoe gebeurt dit?
Als de basis van amyotrofische laterale sclerose een mutatie is in het gen superoxide dismutase-1, ziet het proces er als volgt uit. Mutant superoxide dismutase-1 hoopt zich op in de mitochondriën van motorneuronen (in de krachtcentrales van de cel). Dit "interfereert" met normaal intracellulair transport, inclusief de beweging van eiwitformaties. Eiwitten combineren zich als het ware aan elkaar en dit veroorzaakt het proces van celdegeneratie.
Indien de oorzaak wordt een overmaat glutamaat, het startmechanisme vernietiging van motorneuronen als volgt: Glutamaat open kanalen in het membraan van neuronen calcium. Calcium snelt de cellen in. Overtollig calcium activeert op zijn beurt intracellulaire enzymen. Enzymen zoals "geknipt" neuronale celstructuur, waarbij een grote hoeveelheid vrije radicalen. En deze vrije radicalen beschadigen de neuronen en leiden geleidelijk tot hun volledige vernietiging.
Aangenomen wordt dat de rol van andere factoren in de ontwikkeling van ALS ook ligt in de initiatie van oxidatie van vrije radicalen.
Samenvattend het bovenstaande blijkt dat bij het onderzoek naar de oorzaken en het mechanisme van de ontwikkeling van amyotrofische laterale sclerose meer onbekenden dan het onderzoek. De ambiguïteit in deze vragen wordt weerspiegeld in de manieren om deze ziekte te behandelen. De behandeling, symptomen en diagnose van deze ziekte zijn te vinden in het volgende artikel.
Educatief programma over neurologie, videopresentatie rond het thema "Amyotrofische laterale sclerose

Bekijk deze video op YouTube


