

Secondo l'Organizzazione Mondiale della Sanità (OMS), oggi l'epilessia è una delle cause più comuni di malattie del sistema nervoso. Un terzo della popolazione identificata come epilessia è una malattia permanente e nel 30% dei casi porta ad un esito fatale durante lo stato epilettico. Ogni quinta persona durante la sua vita soffre di almeno un attacco epilettico.
contenuto
- 1Concetti di base
- 2Fattori di rischio per l'epilessia
- 3Convulsioni generalizzate
- 4Convulsioni parziali
-
5Epilessia idiopatica
- 5.1Epilessia assenza bambini.
- 5.2Epilessia giovanile assena.
- 5.3Sindrome di Yants (epilessia mioclonica giovanile).
- 5.4Epilessia con attacchi convulsivi generalizzati.
- 5.5Crampi familiari benigni neonatali.
- 5.6Epilessia rolandica (epilessia pediatrica benigna con punte localizzate centralmente).
- 5.7Epilessia da bambini con parossismi occipitali.
-
6Epilessia sintomatica
- 6.1Epilessia Kozhevnikovskaya.
- 6.2Epilessia del lobo frontale.
- 6.3Epilessia del lobo temporale.
- 6.4Epilessia oscura.
- 6.5Epilessia occipitale.
- 6.6Spasmi infantili.
- 6.7La sindrome di Lenox-Gastaut.
Concetti di base
 L'attacco di epilessia si verifica a causa di un aumento dell'attività elettrica delle cellule nervose.
L'attacco di epilessia si verifica a causa di un aumento dell'attività elettrica delle cellule nervose.L'epilessia è una malattia cronica del sistema nervoso, caratterizzata da ripetuti attacchi epilettici sotto forma di manifestazioni motorie, sensoriali, vegetative e psichiche.
Il sequestro epilettico (attacco) è una condizione transitoria che deriva dallo sviluppo dell'attività elettrica del nervoso cellule (neuroni) nel cervello e si manifesta con sintomi clinici e paraclinici, a seconda della localizzazione dello scoppio scarico.
Al centro di qualsiasi adattamento epilettico vi è lo sviluppo di un'attività elettrica anomala delle cellule nervose che formano una scarica. Se la scarica non va al di là della sua concentrazione, o si diffonde alle parti vicine del cervello, incontra resistenza ed è estinta, allora in questi casi si sviluppano crisi parziali (locali). Nel caso in cui l'attività elettrica si impossessa di tutti i reparti del sistema nervoso centrale, si sviluppa un attacco generalizzato.
Nel 1989 è stata adottata la classificazione internazionale delle epilessie e delle sindromi epilettiche, secondo cui l'epilessia è divisa per il tipo di attacco e il fattore eziologico.
- Epilessia locale (focale, localizzata, parziale):
- Idiopatico (epilessia rolandica, lettura dell'epilessia, ecc.).
- Sintomatica (epilessia Kozhevnikova, ecc.) E criptogenetica.
Questo tipo di epilessia è posto solo se il carattere locale dei parossismi (convulsioni) viene rivelato durante l'esame e l'ottenimento dei dati dell'elettroencefalogramma (EEG).
- Epilessie generalizzate:
- Idiopatico (convulsioni familiari benigne di neonati, epilessia assente da bambino, ecc.).
- Sintomatica (sindrome di West, sindrome di Lennox-Gastaut, epilessia con crisi miocloniche-astatiche, con assenze miocloniche) e criptogenetica.
Gli attacchi con questa forma di epilessia fin dall'inizio sono generalizzati, il che è anche confermato dall'esame clinico e dai dati EEG.
- Epilessie non deterministiche (crampi neonatali, sindrome di Landau-Kleffner, ecc.).
Le manifestazioni cliniche e i cambiamenti nell'EEG hanno caratteristiche sia di epilessia locale che generalizzata.
- Sindromi speciali (convulsioni febbrili, convulsioni che si verificano in disturbi metabolici o tossici acuti).
L'epilessia è considerata idiopatica, se una causa esterna non è stata stabilita durante l'esame, quindi è ancora considerata ereditaria. L'epilessia sintomatica si manifesta quando si riscontrano cambiamenti strutturali nel cervello e malattie, il cui ruolo nello sviluppo dell'epilessia è dimostrato. Le epilessie criptogeniche sono epilessie, la cui causa non è stata identificata e il fattore ereditario è assente.
Fattori di rischio per l'epilessia
- Anamnesi pesata ereditaria (il ruolo dei fattori genetici nello sviluppo dell'epilessia è stato stabilito in modo affidabile);
- Danno cerebrale organico (infezione fetale intrauterina, asfissia intranatale, infezioni postnatali, traumi cerebrali, esposizione a sostanze tossiche);
- Disturbi funzionali del cervello (violazione del sonno / veglia);
- Cambiamenti nell'elettroencefalogramma (EEG);
- Convulsioni febbrili nell'infanzia.
Il quadro clinico di ogni epilessia consiste in convulsioni, che nelle loro manifestazioni sono diverse. Esistono due tipi principali di convulsioni: generalizzata e parziale (motoria o focale).
Per non descrivere ripetutamente il quadro clinico delle stesse crisi con diversi tipi di epilessia, considerali all'inizio.
Convulsioni generalizzate
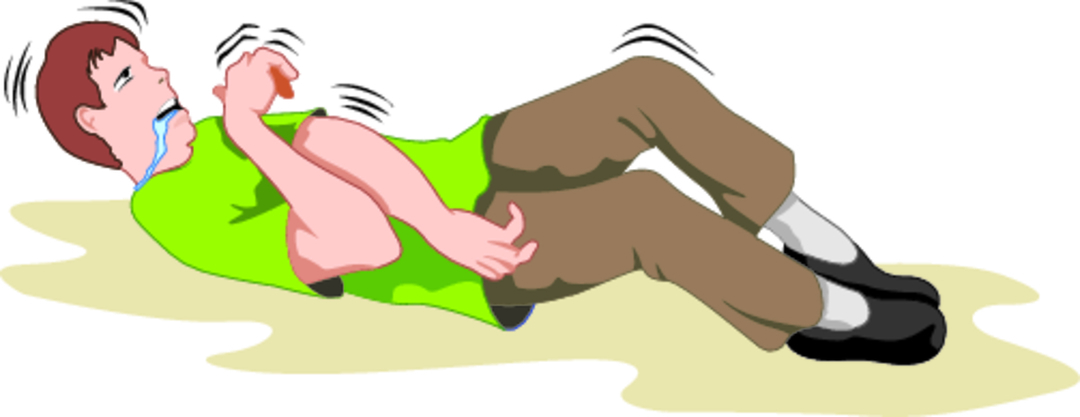 Al momento dell'epilessia generalizzata, una persona perde conoscenza, cade, compaiono convulsioni toniche e cloniche dei muscoli di tutto il corpo.
Al momento dell'epilessia generalizzata, una persona perde conoscenza, cade, compaiono convulsioni toniche e cloniche dei muscoli di tutto il corpo.- Convulsioni tonico-cloniche generalizzate.
Cominciano con una perdita di coscienza, caduta e arcuato allungamento del corpo, quindi si uniscono ai crampi di tutto il corpo. Una persona alza gli occhi, le pupille si allargano, sorge un grido. A causa di uno spasmo convulsivo, l'apnea si sviluppa per alcuni secondi (arresto respiratorio) per diversi secondi, quindi il paziente diventa blu (cianosi). C'è una maggiore salivazione, che in alcuni casi avviene sotto forma di schiuma insanguinata a causa del morso della lingua, minzione involontaria. Durante un attacco, quando cadi privo di sensi, puoi subire un grave infortunio. Dopo un attacco, una persona di solito si addormenta o diventa pigra, rotta (periodo post-ammissione).
Questo tipo di parossismi (convulsioni) si verifica spesso con forme ereditarie di epilessia o sullo sfondo di danni cerebrali tossici all'alcol.
- Convulsioni miocloniche.
Le mioclonie sono contrazioni muscolari a scatti per alcuni secondi, possono essere ritmiche o irregolari. Per questo tipo di convulsioni, è tipica la contrazione muscolare, che può colpire singole parti del corpo (viso, braccio, tronco) o essere generalizzata (in tutto il corpo). Nella clinica, questi attacchi sembreranno una scrollata di spalle, un movimento della mano, squat, spremere i pennelli, ecc. La coscienza è spesso preservata. Si verificano nella maggior parte dei casi durante l'infanzia.
- Assenze.
Questo tipo di convulsione avviene senza convulsioni, ma con una breve deenergia momentanea della coscienza. Una persona diventa come una statua con gli occhi aperti e vuoti, non entra in contatto, non risponde alle domande e non reagisce agli altri. L'attacco dura in media 5 secondi. fino a 20 secondi, dopo di che una persona viene da sé e continua le attività interrotte. A proposito dell'attacco non ricorda nulla. Le cadute durante le assenze tipiche non sono caratteristiche del paziente. Le assenze di breve durata possono passare inosservate dalla persona stessa o dalle persone che lo circondano. Più spesso questo tipo di attacchi è osservato nelle epilessie idiopatiche nei bambini da 3 a 15 anni. Negli adulti, di norma, non iniziano.
Ci sono anche assenze atipiche, che sono più lunghe nel tempo e possono essere accompagnate da cadute nella persona e dalla minzione involontaria. Si verificano principalmente nell'infanzia con epilessie sintomatiche (danno cerebrale organico grave) e sono combinati con un disturbo della psiche e dello sviluppo intellettuale.
- Attacchi o crisi achetiche.
L'uomo perde bruscamente il tono, provocando una caduta, che spesso porta a ferite alla testa. Potrebbe esserci una perdita di tono in parti separate del corpo (penzoloni della testa, penzoloni della mascella inferiore). L'assenza può anche essere una perdita di tono, ma si verifica più lentamente (la persona si assesta), e in questo caso si tratta di una goccia acuta e precipitosa. Le convulsioni atoniche durano fino a 1 minuto.
Convulsioni parziali
- Una semplice misura parziale.
Questo gruppo include parossismi motori, sensoriali, autonomi e psichici. Le manifestazioni dipendono dalla localizzazione del focus epilettico nel cervello. Ad esempio, gli attacchi motori si manifestano con spasmi, crampi muscolari, rotazioni del busto e testa, suono o, al contrario, interruzione della conversazione quando la messa a fuoco si trova nell'area del motore CNS. Inoltre ci sono movimenti masticatori, labbra schioccanti, labbra leccate.
Le convulsioni sensoriali (area sensoriale nel lobo parietale) sono caratterizzate da attacchi locali di parestesia (gonfiore, formicolio), intorpidimento di parte del corpo, sensazione di sapore sgradevole (amaro, salato) o odore, disturbi visivi sotto forma di "scintille davanti agli occhi" o perdita di campi visivi, flash (temporale e lobo occipitale).
Le convulsioni autonome si manifestano sotto forma di colorazione alterata della pelle (scottature o arrossamenti), frequenti battito cardiaco, aumento o diminuzione della pressione sanguigna, cambiamenti nella pupilla, comparsa di sensazioni spiacevoli nell'epigastrico zona.
Le convulsioni mentali sono una sensazione di paura, un afflusso di pensieri, un cambiamento nel linguaggio, un sentimento di ciò che è già stato visto o già sentito, l'apparenza di una sensazione di irrealtà di ciò che sta accadendo. Oggetti e parti del corpo possono apparire alterati per forma e dimensioni (ad esempio, la gamba sembra piccola e il braccio è enorme). Le crisi mentali sono raramente indipendenti, spesso precedono complesse crisi parziali.
Durante semplici crisi parziali, la coscienza non è persa.
- Sequestro parziale complicato.
È un semplice attacco parziale, ma vi si unirà una violazione della coscienza. Una persona realizza il suo attacco, ma non può entrare in contatto con le persone circostanti. Tutti gli eventi che si verificano con una persona durante un attacco vengono amnesiati (dimenticati) da lui stesso. Le funzioni cognitive umane sono disturbate - c'è una sensazione di irrealtà di ciò che sta accadendo, nuovi cambiamenti dentro di sé.
- Convulsioni parziali con generalizzazione secondaria.
Iniziare con crisi parziali semplici o complesse, quindi passare a convulsioni tonico-cloniche generalizzate. Dura fino a 3 minuti, dopo di che una persona spesso si addormenta.
Epilessia idiopatica
 Con l'epilessia idiopatica, non vengono rilevati cambiamenti anormali del cervello sulla risonanza magnetica.
Con l'epilessia idiopatica, non vengono rilevati cambiamenti anormali del cervello sulla risonanza magnetica.Questa specie è, di regola, di natura ereditaria e si manifesta nell'infanzia, nell'adolescenza o nell'adolescenza. Il decorso delle principali varianti dell'epilessia idiopatica è benigno e raramente è accompagnato da disturbi mentali e altri disturbi neurologici. Nella diagnosi, i cambiamenti nell'EEG sono rilevati solo durante il periodo di crisi. Su SCT e MRI, la patologia del cervello non è determinata. Una buona risposta è data dall'epilessia alla terapia anticonvulsivante e ad un'alta percentuale di remissioni spontanee (recuperi).
Di seguito sono riportati i principali tipi di epilessia idiopatica:
Epilessia assenza bambini.
La malattia inizia all'età di 4 anni a 10 anni con assenze semplici (a breve termine mute, bambino non entra in contatto, non risponde alle domande e non reagisce agli altri), che vengono ripetute molte volte giorno. In una parte dei bambini, convulsioni generalizzate tonico-cloniche (perdita di coscienza, convulsioni di tutto il corpo) si uniscono alle assenze.
Epilessia giovanile assena.
In metà dei casi, fa il suo debutto con convulsioni tonico-cloniche generalizzate (vedi fig. sopra), e nella restante metà della malattia inizia con semplici assenze. Nel 10% di pazienti nell'anamnesi ci sono convulsioni febbrili (le convulsioni con un aumento di temperatura del corpo). Di regola, c'è un singolo attacco con un intervallo di 2-3 giorni. Può essere trattato con valproato.
-
.
Sindrome di Yants (epilessia mioclonica giovanile).
Una forma comune di epilessia, che viene spesso confusa con l'epilessia frontale. Nella metà dei casi inizia nell'adolescenza. La clinica è rappresentata da varie mioclonie (spasmi muscolari), più spesso nei muscoli delle braccia e della fascia scapolare. Le contrazioni possono essere di diversa gravità, da appena evidenti a pronunciate. Dopo un po 'dall'esordio della malattia, possono associarsi convulsioni generalizzate. La frequenza delle crisi varia da un caso all'altro in diverse settimane. Provocare un attacco può essere un violento risveglio, un senso di ansia, insonnia, assunzione di alcool o improvvisi e forti stimoli. La diagnosi viene fatta sulla base dell'esame clinico e dei dati EEG. L'alta frequenza delle recidive distingue questa forma di epilessia dagli altri.
Epilessia con attacchi convulsivi generalizzati.
L'insorgenza della malattia cade anche nell'adolescenza. La frequenza delle crisi varia in un ampio intervallo (da una volta alla settimana a una volta all'anno). La malattia è geneticamente determinata. In alcuni casi, assenze o crisi miocloniche possono unirsi alle crisi tonico-cloniche generalizzate. Il trattamento è iniziato con valproato o carbamazepina. Con una terapia opportunamente selezionata, la remissione si verifica nel 90% dei pazienti.
Crampi familiari benigni neonatali.
La malattia è di natura familiare, ereditata da un tipo autosomico dominante ed è estremamente rara. I primi sintomi compaiono nel periodo neonatale del bambino (fino a 28 giorni di vita). Le mamme osservano le contrazioni convulsive dei muscoli del corpo del bambino, in alcuni bambini può esserci un'apnea a breve termine (arresto della respirazione). Il decorso della malattia è favorevole, si sviluppano convulsioni tonico-cloniche raramente generalizzate. L'appuntamento di terapia antiepilettica a lungo termine non è razionale, poiché all'età di un anno del bambino, nella maggior parte dei casi, arriva una remissione indipendente.
-
.
Epilessia rolandica (epilessia pediatrica benigna con punte localizzate centralmente).
Il debutto della malattia cade nell'età dei bambini (2-10 anni) nell'80% dei casi. Il quadro clinico è rappresentato da un semplice motore (crampi muscolari facciali, spasmi della lingua, disturbi del linguaggio, comparsa di una voce roca, respiro sibilante, ecc.), Sensoriale (sensazioni di formicolio sul viso) o attacchi vegetativi (faccia rossa, palpitazioni), che nella maggior parte dei casi compaiono di notte (durante il periodo di addormentarsi e il risveglio). A volte ci sono crisi parziali complesse (vedi Fig. sopra) e raramente convulsioni generalizzate secondarie (vedi Fig. sopra). L'attacco è preceduto da un'aura: una sensazione di formicolio, una corrente elettrica, un "gozzo strisciante". Il sequestro stesso è una contrazione di metà dei muscoli facciali o strappi convulsivi delle labbra, metà della lingua, faringe. Durante un attacco, la salivazione aumenta, la voce scompare, ci sono suoni rauca strizzati. In alcuni casi, le convulsioni possono diffondersi al braccio sullo stesso lato del corpo e, molto raramente, al piede. L'attacco dura non più di 2 minuti. Il decorso della malattia è benigno: in alcuni bambini in tutta la vita può esserci solo un episodio di crisi parziali e in alcuni bambini oltre 13 anni di convulsioni possono interrompere senza farmaci senza anticonvulsivanti (anticonvulsivanti).
Epilessia da bambini con parossismi occipitali.
Il focus epilettico è nel lobo occipitale, quindi le funzioni visive sono interrotte (perdita di campi visivi, lampi di luce davanti agli occhi). Il secondo sintomo sono i mal di testa, che sono di natura emicranica. Questa forma di epilessia ha due origini: esordio precoce (1-3 anni) e successiva (3-15 anni). Con il debutto precoce della malattia nella clinica ci sono gravi attacchi che iniziano al momento il risveglio del bambino e sono accompagnati da forte vomito, mal di testa, poi c'è un giro della testa dentro lato. L'attacco termina con convulsioni o convulsioni generalizzate in una metà del corpo (emiconvulsioni). Durante il sequestro, la coscienza è assente.
Con l'insorgenza tardiva della malattia, si osservano semplici crisi sensoriali in una persona (allucinazioni visive sotto forma di piccole figure multicolori sulla periferia dei campi visivi). Il sequestro dura in media 1-3 minuti, quindi viene sostituito da un forte mal di testa, nausea e vomito.
Nel trattamento delle droghe di scelta sono anche valproato. La diagnosi si basa sui dati EEG e sull'esame clinico. Il corso è favorevole e la remissione avviene nell'80% o più di bambini.
Epilessia sintomatica
 Il trauma craniocerebrale può causare lo sviluppo dell'epilessia sintomatica.
Il trauma craniocerebrale può causare lo sviluppo dell'epilessia sintomatica.Le cause di questa forma di epilessia includono lesioni craniocerebrali, tumori, traumi alla nascita, danno cerebrale tossico-infettivo, malformazioni artero-venose, ecc.
I sintomi dipendono dalla posizione topica del focus epilettico nel cervello. Esistono i seguenti tipi di epilessia sintomatica:
Epilessia Kozhevnikovskaya.
È un semplice attacco del motore: convulsioni nella mano o nel viso, la cui intensità può variare. Una caratteristica speciale di Kozhevnikov epilepsii- durata attacco, che può durare un paio di giorni e lascia una paresi (debolezza) quella parte del corpo, che si contraevano. In un sogno, la contrazione può diminuire o scomparire. Di norma, solo un lato del corpo è coinvolto nel processo (braccio sinistro e gamba sinistra, metà destra della faccia, ecc.). Le assenze con epilessia kozhevnikovskoy non sono trovate.
Nei bambini, la malattia si verifica più spesso dopo l'encefalite di un'eziologia sconosciuta. Il trattamento è principalmente finalizzato all'eliminazione della causa e dello scopo degli anticonvulsivanti. Per questo tipo di epilessia, esiste anche un metodo chirurgico di trattamento, che viene utilizzato per l'inefficacia della terapia conservativa e delle malattie gravi.
Epilessia del lobo frontale.
Una delle più diverse manifestazioni cliniche di epilessia. Caratteristica dell'inizio improvviso di un attacco, che dura non più di 30 secondi. e finisce anche all'improvviso. L'attacco rappresenta movimenti eccessivi, gesti. A volte prima dell'attacco una persona può provare una sensazione di calore, ragnatele sul corpo. Colorazione emotiva caratteristica delle convulsioni, può essere la minzione involontaria. L'epilessia frontale si manifesta spesso come semplice e complessa (cfr. sopra) convulsioni parziali, nonché convulsioni miocloniche (spasmi di alcuni gruppi muscolari). Le convulsioni tendono a generalizzare il processo e a formare uno stato epilettico, si verificano più spesso durante la notte.
Epilessia del lobo temporale.
La natura delle convulsioni può essere diversa - dal semplice motore (afferrando gli altri, grattandosi, crampi al braccio o alla gamba) al secondario generalizzato. Appare a qualsiasi età. La persona è turbata dalle illusioni, una sensazione di irrealtà di ciò che sta accadendo, poiché l'attenzione è nel lobo temporale, quindi può apparire olfattivo e assaggiare le allucinazioni (una sensazione salata nella lingua, un senso dell'olfatto rot). C'è uno sguardo rigido, gli automatismi ororalimentatnye (schiocco delle labbra, labbra leccate, lingua sporgente), paura, panico. Il timbro della voce può cambiare, appare un discorso incoerente. L'attacco dura circa 2 minuti. Nel trattamento di farmaci di scelta sono valproato e carbamazepina. Nel 30% dei casi, l'epilessia non risponde al trattamento con farmaci antiepilettici.
Epilessia oscura.
Una causa comune dell'epilessia parietale sono i tumori e il trauma craniocerebrale (conseguenze). Una persona è preoccupata per le convulsioni sensoriali che sono polimorfiche nelle manifestazioni cliniche (sensazioni di freddo, bruciore, prurito, formicolio, intorpidimento, ecc.), allucinazioni (non sente il suo corpo, la sensazione di una disposizione impropria di parti del corpo, ad esempio, la mano "esce" da addome). Durante un attacco, il disorientamento si verifica nello spazio se l'attenzione epilettica è nell'emisfero non dominante. Il sequestro dura non più di 2 minuti, ma può essere ripetuto più volte, più volte al giorno. Il trattamento è principalmente finalizzato all'eliminazione della causa della malattia.
Epilessia occipitale.
Le cause sono anche diverse (malformazione artero-venosa, tumore, trauma). Nel lobo occipitale c'è una zona dell'analizzatore visivo, quindi nella clinica dell'epilessia occipitale ci sono attacchi semplici sotto forma di deficit visivo fino alla cecità, flash, allucinazioni visive, perdita di campi view. Abbastanza spesso le crisi parziali semplici diventano convulsioni generalizzate. In alcuni casi, potrebbe esserci un tremito delle palpebre, girando la testa e gli occhi di lato. Nel periodo post-fatale c'è un mal di testa, un disturbo generale. Nel trattamento, vengono utilizzati anticonvulsivanti.
Spasmi infantili.
La malattia si verifica nei bambini del primo anno di vita sullo sfondo di malformazioni del cervello, trauma della nascita, Il quadro clinico è rappresentato dalla contrazione dei muscoli del tronco, che vanno in gruppi. Durante l'attacco, il bambino gira la testa, piega le braccia e le gambe, si piega "come un temperino" a causa delle contrazioni dei muscoli addominali. Il ritardo mentale si sviluppa.
La sindrome di Lenox-Gastaut.
Le cause che portano a questa malattia sono diverse (encefalite, meningite, anomalie dello sviluppo del cervello). La sindrome si manifesta nell'età da 1 anno a 5 anni. Le convulsioni sono assenze, attacchi atonici (forte flessione delle gambe nelle articolazioni del ginocchio, taglienti testa cadente o rapida perdita di oggetti dalle mani) e convulsioni miocloniche (contrazioni nei flessori mani). Sviluppa un ritardo nello sviluppo psico-motorio. Nella diagnosi, l'EEG, la RM e la SCT vengono utilizzati insieme ad un esame clinico da uno specialista.
La varietà di forme cliniche di epilessia, così come la diversa manifestazione della stessa crisi in persone diverse (ad esempio, intensità diversa convulsioni, la loro frequenza, la risposta alla terapia) causano notevoli difficoltà nella diagnosi e nella selezione di adeguati anticonvulsivanti la terapia. Nonostante questo, oggi nell'arsenale del dottore ci sono moderni metodi diagnostici (EEG, MRI, SCT, ecc.) che facilitano il processo di stabilire una diagnosi definitiva, così come le medicine di nuovo generazioni.
Un video cognitivo su "Quali tipi di attacchi esistono

Guarda questo video su YouTube
Canale 7, un medico neuropatologo parla delle cause e dei tipi di epilessia:

Guarda questo video su YouTube
Programma educativo sulla neurologia, lezione sul tema "Classificazione dell'epilessia

Guarda questo video su YouTube


